Bioinformatics analysis of sequencing data
Bioinformatics analysis of sequencing data is the main stage when a medical report is formed. The patient is searched for pathogenic and likely pathogenic variants in accordance with the original referral diagnosis, as well as similar phenotypic features.
Данные секвенирования анализируются с помощью автоматизированного алгоритма, заключающего в себя оценку параметров качества секвенирования.
Data analysis. Используется автоматизированный пайплайн, написанный на языке python, включающий в себя оценку качества секвенирования (FastQC), удаление оснований и ридов с низким качеством (fastp), выравнивание прочтений на референсную сборку генома человека GRCh38/hg38 при помощи bwa-mem2, коллинг вариантов с помощью strelka2. Метрики покрытия получены программой Picard. Аннотация полученных вариантов реализована при помощи OMIM и VEP. Анализ числа копий (инсерций и делеций) проводился с помощью clinCNV и manta, интерпретация в соответствии с критериями ACMG3.
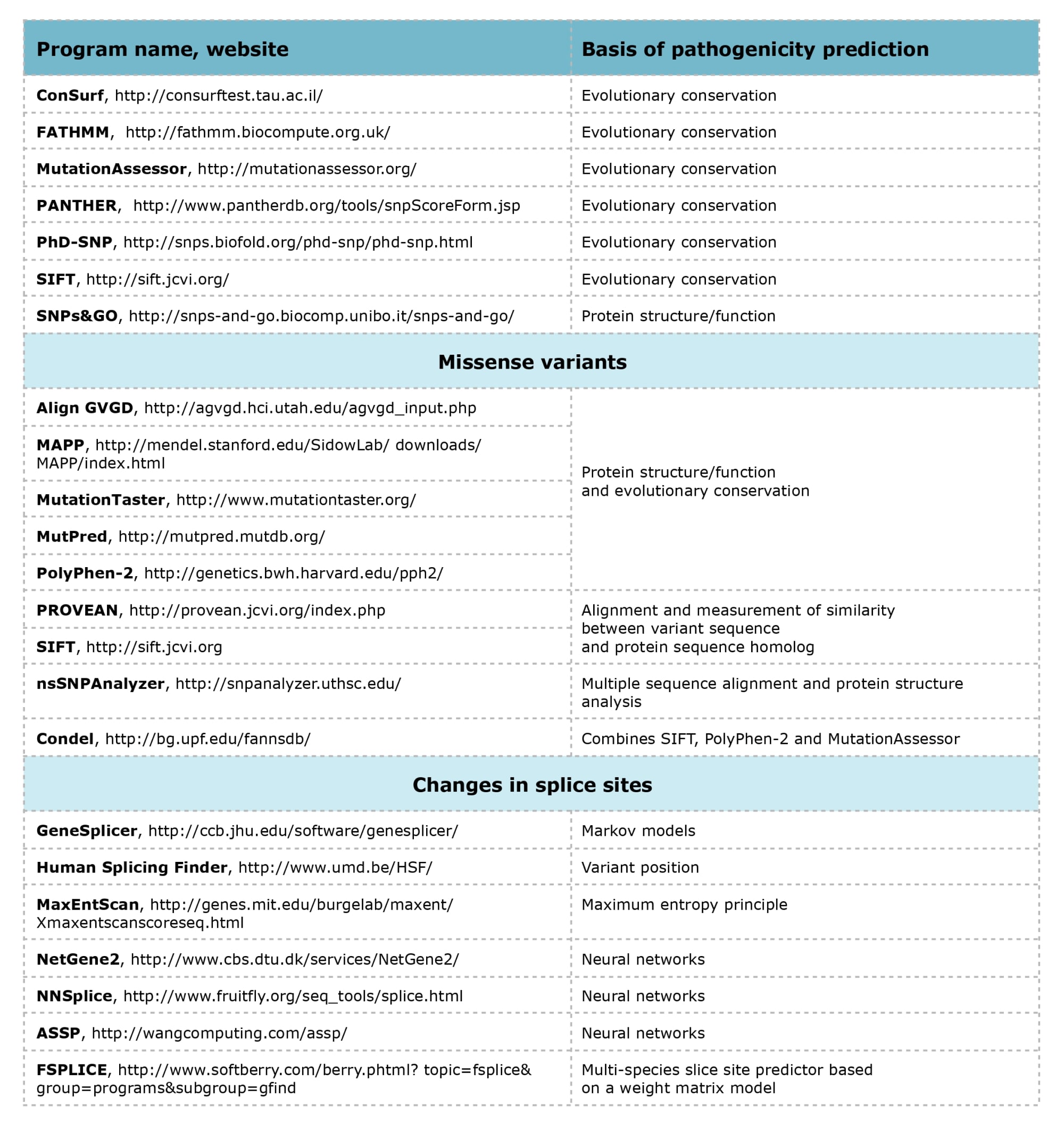
Computer programs used for predicting the pathogenicity of nucleotide sequence variants
When conducting the analysis, we strictly follow Russian and international recommendations that can be found at the following links:
- https://www.medgen-journal.ru/jour/article/view/642/402
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4544753/
Recommended databases for analyzing the pathogenicity of nucleotide sequence variants

Technology stack used:
- Python
- Docker
- Snakemake
- ClickHouse
- MongoDB
General requirements for the preparation of a bioinformatics report and conclusion on the results of the study
Information about the patient should include last name, first name, patronymic if there is (in three lines below each other), date of birth, gender, clinical diagnosis, indications of family relationships if there is information about other family members.
Information on identified nucleotide sequence variants should include:
a list of pathogenic, likely pathogenic and variants of uncertain significance identified during the analysis, according to the HGVS nomenclature;
a gene name;
a position of the substitution in the genome according to GRCh/hg;
zygosity;
an exon number;
a nomenclature at nucleotide and DNA level;
a protein-level nomenclature (in cases where historical (alternative) nomenclature is used, iit is recommended to specify both variants);
a number of the reference sequence used (NM);
coverage details:
- среднее покрытие исследуемой области при секвенировании полного экзома (WES) должно быть не менее х70, полного генома (WGS) ‒ не менее х30;
- when studying WES/WGS/"clinical" exome, it is necessary to indicate the proportion (in %) of «target» areas with coverage less than x10;
- when studying gene panels, it is necessary to indicate all regions with coverage less than x10;
- if only one variant is detected in a heterozygous state in a disease with a recessive type of inheritance, it is necessary to indicate all gene regions with coverage of less than x10 for any type of study.
a name of the disease;
a type of inheritance;
a frequency of the variant in databases (indicating the database version used);
results of in silico pathogenicity prediction programs indicating the Score and the program version used. The results of the analysis of the pathogenicity of the variant using all the programs that the laboratory uses to interpret its clinical significance should be reported, not just those results where it was found to be clinically significant.
All nucleotide sequence variants enumerated in the report must be confirmed by Sanger sequencing or other validated technology.