ПГТ. Биоинформатический анализ
BAM файл, полученный с помощью ПО «Torrent Suite», передается в ПО набора «Ion Reporter». Выявление анеуплоидии выполняется с использованием алгоритма на основе скрытой марковской модели Hidden Markov Model (HММ).
Скрытая марковская модель позволяют для заданной модели λ = (A, B, π). и последовательности O = {o1, …oT} подсчитать вероятность P(O/λ) порождения последовательности наблюдения O = {o1, …oT} моделью λ и наиболее вероятную последовательность Q= {q1, …qT}. Пусть даны последовательность наблюдений O = {o1, …oT} и модель λ = (A, B, π). Алгоритм Витерби используется для того, чтобы выбрать последовательность состояний Q= {q1, …qT}, которая с наибольшей вероятностью для данной модели P(O/λ) генерирует последовательность наблюдений O = {o1, …oT}. В скрытой марковской модели можно следить лишь за переменными, на которые оказывает влияние данное состояние. Каждое состояние имеет вероятностное распределение среди всех возможных выходных значений. Поэтому последовательность символов, сгенерированная НММ, даёт информацию о последовательности состояний.

Овалы представляют собой переменные со случайным значением. Случайная переменная x(t) представляет собой значение скрытой переменной в момент времени t. Случайная переменная y(t) — это значение наблюдаемой переменной в момент времени t. Стрелки символизируют условные зависимости. Значение скрытой переменной x(t) (в момент времени t ) зависит только от значения скрытой переменной x(t-1) (в момент t-1). Это называется свойством Маркова. Хотя в то же время значение наблюдаемой переменной y(t) зависит только от значения скрытой переменной x(t) (обе в момент времени t).
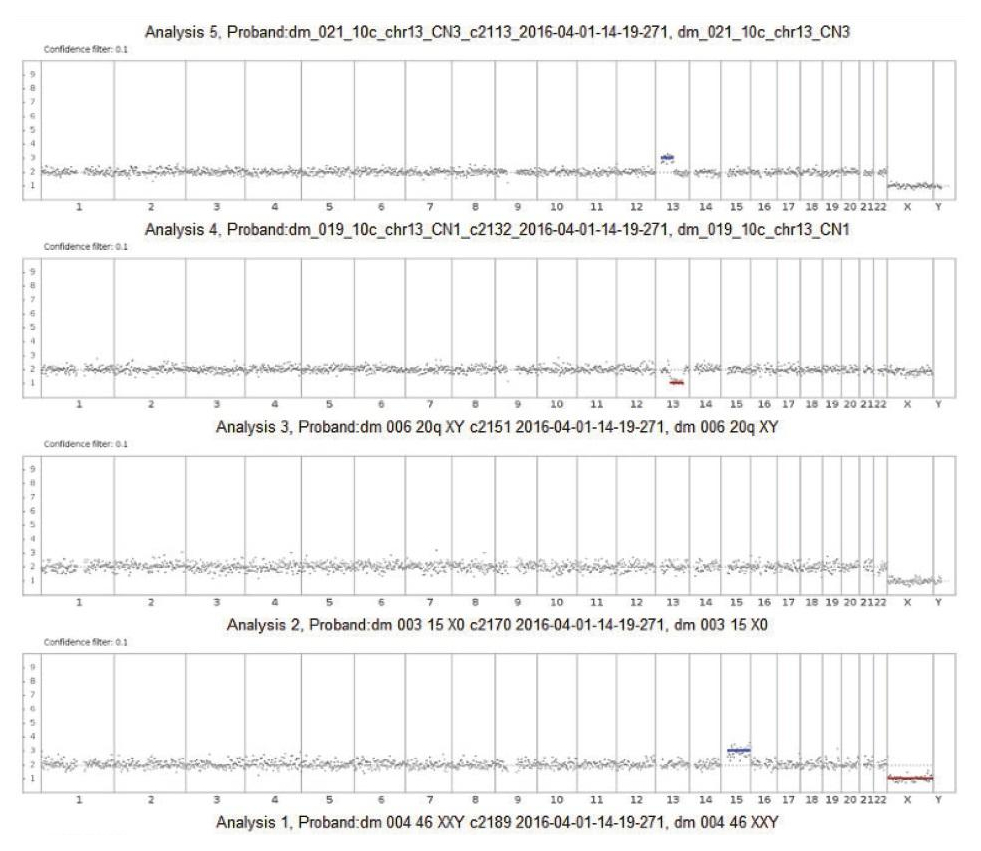
Алгоритм использует статистическую модель при анализе прочтений, покрывающих весь геном для предсказания числа копий хромосом. Перед определением числа копий, прочтения корректируются на предмет ошибок GC участков и сравниваются с предварительно установленной базовой линией, путем анализа 10 нормальных мужских образцов тем же способом.
Использование 10 образцов для расчета базовой линии существенно уменьшает вариабельность покрытия от образца к образцу и приводит к меньшему количеству ложноположительных отметок. Использование заданной линии было валидировано на образцах при межлабораторном сравнении.
Использование HММ позволяет статистически обработать всю необходимую информацию по образцу для определения вероятности отклонения конкретной области генома от заданного значения плоидности. Анализ данных позволяет обеспечить настраиваемые коэффициенты обсчёта. Параметры алгоритма настроены на обработку данных полногеномного секвенирования с низким покрытием с использованием технических (стандартных) образцов анализируемых в двух повторах, а также образцов с известными анеуплоидиями. Изменение метрик ПО позволяет выбирать уровень чувствительности (низкая, средняя и высокая). Протокол с высокой чувствительностью позволяет анализировать хромосомные сегменты, а также выявлять анеуплоидии в шумных регионах, но при этом увеличивая вероятность ложноположительного результата. Средняя чувствительность — параметр, используемый по умолчанию, и рекомендуемый для использования как первичный. Протокол с низкой чувствительностью является наиболее «требовательным» и обнаруживает только регионы с крайне высокой достоверностью наличия анеуплоидии.