Биоинформатический анализ данных секвенирования
Биоинформатический анализ данных секвенирования — основной этап на которому формируется Заключение. У пациента проводится поиск патогенных и вероятно патогенных вариантов в соответствии с исходным направительным диагнозом, а также схожими фенотипическими признаками.
Данные секвенирования анализируются с помощью автоматизированного алгоритма, заключающего в себя оценку параметров качества секвенирования.
Анализ данных. Используется автоматизированный пайплайн, написанный на языке python, включающий в себя оценку качества секвенирования (FastQC), удаление оснований и ридов с низким качеством (fastp), выравнивание прочтений на референсную сборку генома человека GRCh38/hg38 при помощи bwa-mem2, коллинг вариантов с помощью strelka2. Метрики покрытия получены программой Picard. Аннотация полученных вариантов реализована при помощи OMIM и VEP. Анализ числа копий (инсерций и делеций) проводился с помощью clinCNV и manta, интерпретация в соответствии с критериями ACMG3.
Используемые компьютерные программы предсказания патогенности вариантов нуклеотидной последовательности
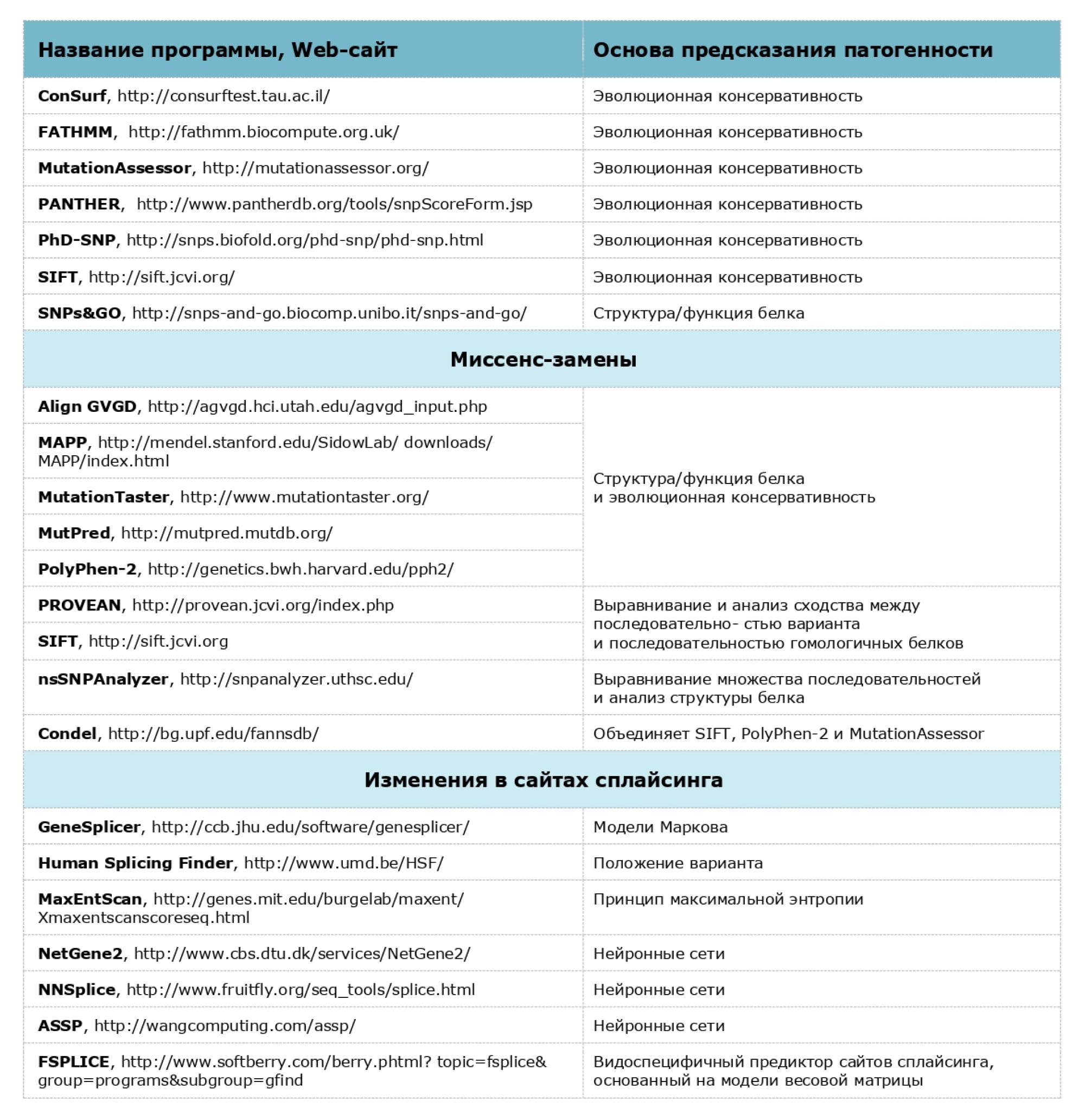
При проведении анализа мы строго следуем российским и международным рекомендациям, ознакомиться с которыми можно по следующим ссылкам:
- https://www.medgen-journal.ru/jour/article/view/642/402
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4544753/
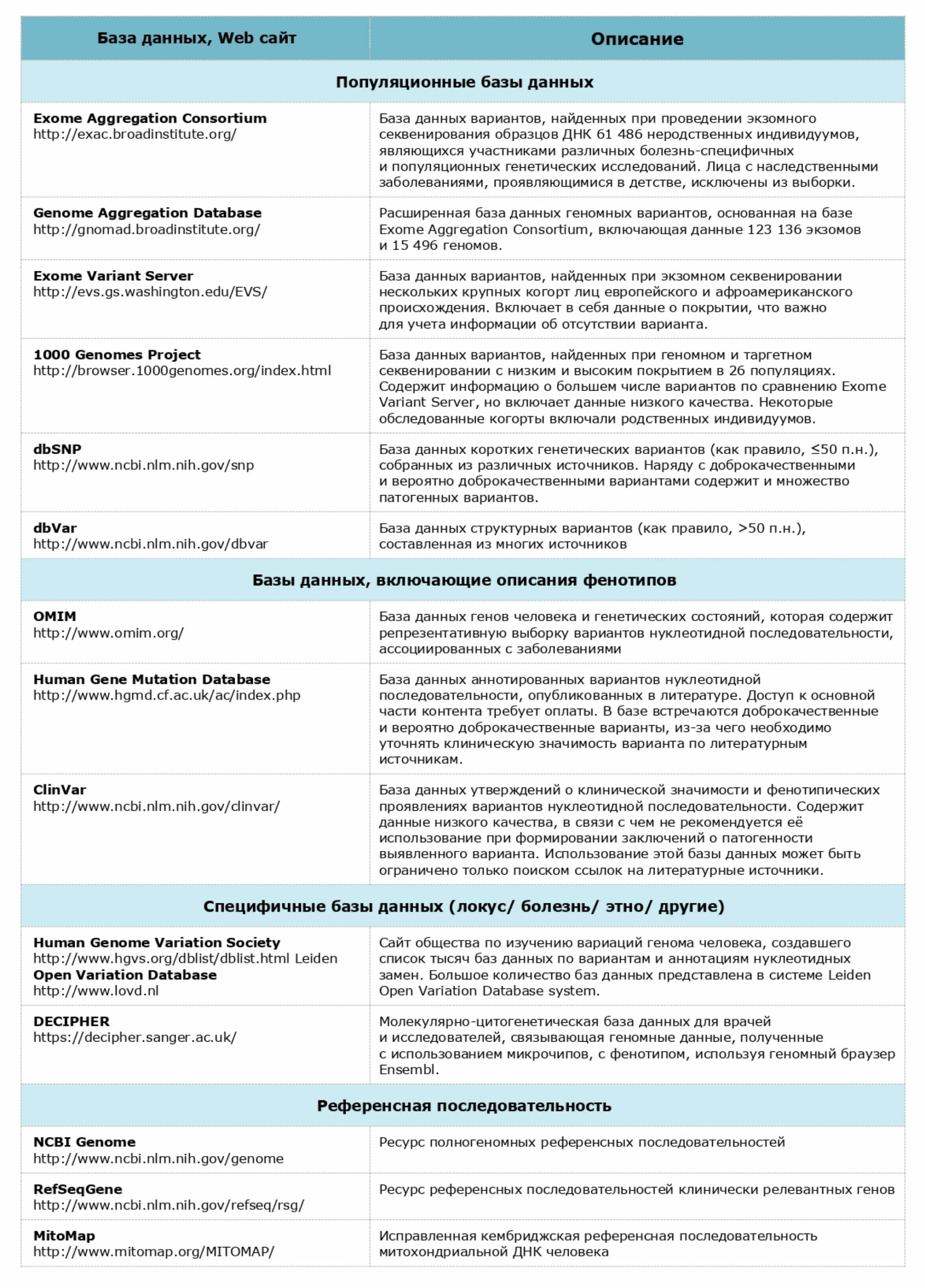
Рекомендуемые базы данных для анализа патогенности вариантов нуклеотидной последовательности
Используемый стек технологий:
- Python
- Docker
- Snakemake
- ClickHouse
- MongoDB
Общие требования к оформлению биоинформатического отчёта и заключения о результатах исследования
Информация о пациенте должна включать фами лию, имя, отчество (в трех строках друг под другом), дату рождения, пол, клинический диагноз, указания на родственные отношения, если есть данные о других членах семьи.
Информация о выявленных вариантах нуклеотидной последовательности должна включать:
список патогенных, вероятно патогенных вариантов и вариантов неопределенного значения, выяв- ленных при анализе, по номенклатуре HGVS;
название гена;
положение (позицию) замены в геноме по GRCh/hg;
зиготность;
номер экзона;
номенклатуру на уровне нуклеотида и ДНК;
номенклатуру на уровне белка (в случаях, когда используется историческая (альтернативная) номенклатура, рекомендуется указывать оба варианта);
номер используемой референсной последова- тельности (NM);
данные о покрытии:
- среднее покрытие исследуемой области при секвенировании полного экзома (WES) должно быть не менее х70, полного генома (WGS) ‒ не менее х30;
- при исследовании WES/WGS/»клинического» экзома необходимо указывать долю (в %) «целе- вых» областей с покрытием менее x10;
- при исследовании панелей генов необходимо указывать все регионы с покрытием менее x10;
- при обнаружении только одного варианта в гетерозиготном состоянии при заболевании с рецессивным типом наследования необходимо указывать все области гена с покрытием менее х10 при любом типе исследования.
название заболевания;
тип наследования;
частоты варианта в базах данных (с указанием использованной версии базы данных);
результаты программ предсказания патогенности in silico с указанием Score и использованной вер- сии программы. Должны быть указаны результаты анализа патогенности варианта с использованием всех программ, которые применяет лаборатория для интерпретации его клинической значимости, а не только те результаты, где он оказался клинически значимым.
Все варианты нуклеотидной последовательности, указанные в заключении, должны быть подтверждены секвенированием по Сэнгеру или другой валидированной технологией.