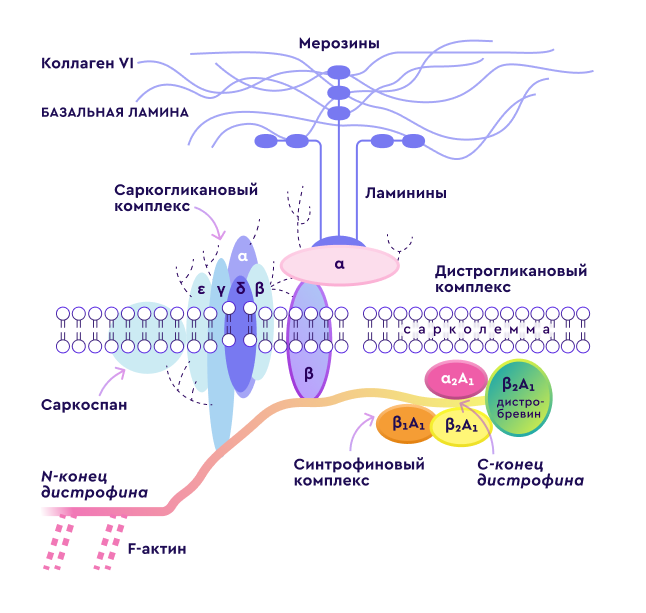
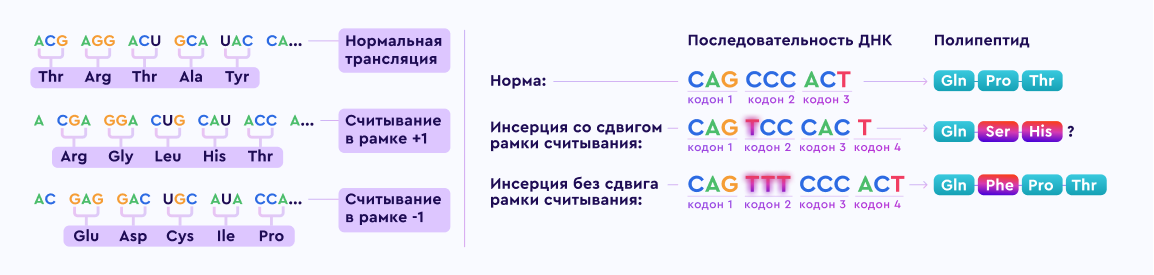
Ген дистрофина локализован на коротком плече Х хромосомы (Xp21.2-p21.1) и является одним из самых протяженных в человеческом геноме. Дистрофинопатии по типу наследования являются Х‑сцепленными рецессивными заболеваниями.
Большинство случаев обусловлены гетерозиготным носительством матерью патологической мутации в гене DMD.
При беременности мальчиком — 50%Мутация будет передана сыну и он будет болен ПМДД/Б.
Все мальчики, унаследовавшие от матери патогенный вариант, будут больны.
При беременности девочкой — 50%Дочь унаследует данную мутацию и станет носительницей дефектнойго копии гена DMD.
Девочки, унаследовавшие патогенный вариант могут быть как бессимптомными носителями патогенной мутации в гетерозиготном состоянии, так и иметь клинические проявления классической дистрофинопатии.
Выявление носительства мутации гена DMD и планирование семьи с учетом риска рождения больного ребенка — самый эффективный способ профилактики дистрофинопатий.
Отец больного мальчика, как правило, не нуждается в молекулярно-генетическом тестировании, поскольку если он здоров, то он не может быть гемизиготным по патогенному варианту.
Остаточный риск — это вероятность рождения мальчика с дистрофинопатией, даже если при исследовании гена DMD у матери носительства патогенного варианта в лейкоцитарной ДНК не обнаружено.
Существование остаточного риска обусловлено тем, что в 15-20% случаев развитие мышечной дистрофии происходит из-за мутации, возникшей de novo (наиболее вероятен гонадный мозаицизм) [5].
В таком случае все братья пробанда-мальчика также имеют повышенный риск унаследовать патогенный вариант и при последующих беременностях целесообразно проведение пренатальной диагностики.
Прогрессирующая мышечная дистрофия Дюшенна:
- Первые признаки проявляются в возрасте до 5 лет.
- Отмечается низкая активность ребёнка.
- Прогрессирующая симметричная мышечная слабость в проксимальных отделах.
- Частые падения, двигательная неловкость, быстрая утомляемость.
- Псевдогипертрофия мышц (увеличение объема при снижении функциональных возможностей), наиболее часто икроножных, может проявляться в форме ложного впечатления атлетического телосложения.
- Распространение патологического процесса носит восходящий характер. Первыми поражаются мышцы нижних конечностей, затем плечевого пояса, спины и проксимальных отделов верхних конечностей.
- На ранних стадиях снижаются сухожильные рефлексы.
- Примерно в возрасте 18 лет развивается кардиомиопатия [8], которая проявляется в виде гипертрофии левого желудочка и аритмии.
- Зависимость от инвалидной коляски развивается до 13 лет.
Прогрессирующая мышечная дистрофия Беккера:
- Дебют в возрасте от 10 до 20 лет с появления слабости и утомляемости мышц тазового пояса и ног.
- Ранними симптомами части становятся мышечные спазмы.
- Клинические проявления сходны с ПМДД, но выражены слабее.
- Прогрессирующая симметричная мышечная слабость в проксимальных отделах; слабость четырехглавой мышцы бедра иногда является единственным признаком развивающейся болезни.
- Зависимоть от инвалидной коляски развивается после 16 лет, хотя некоторые пациенты сохраняют самостоятельную двигательную активность в 30 и в редких случаях в 40 лет.
- В ряде случаев выявляются гипогенитализм и атрофия яичек [9].
Методы молекулярно-генетической диагностики
Подход к молекулярно-генетической диагностике дистрофинопатий заключается в последовательном проведении следующих этапов:
Исследование гена DMD
Так как большинство случаев ПМДД/ПМДБ вызвано потерей или удвоением протяженных участков ДНК, целесообразно начать с поиска мутаций методом MLPA (хромосомный микроматричный анализ экзонного уровня).
Если патогенный вариант не обнаружился, то следующим шагом проводится секвенирование гена DMD.
Исследование панели генов
Мутации которых характерны для нервно-мышечных заболеваний, в том числе со сходными клиническими проявлениями.
Расширенный генетический анализ
Включающий полноэкзомное или полногеномное секвенирование может быть проведен в случае нетипичных клинических проявлений, с целью уточнения диагноза и установления возможных причинных находок в других генах.
Как правило, исследование проводится с учетом информации о ранее выявленных патогенных вариантах в гене DMD у членов семьи.
Биологические образцы, пригодные для проведения молекулярно-генетического исследования:
- Периферическая кровь (пробирка с ЭДТА)
- Околоплодные воды, взятые на 16 неделе беременности
- Ворсины хориона
- Пуповинная кровь (пробирка с ЭДТА)