Специалисты
Многолетний опыт работы в области генетики, лабораторной диагностики и биоинформатики
Поиск делеций и дупликаций в гене дистрофина
Клиническая характеристика
Дистрофинопатии представляют собой группу Х‑сцепленных нервно‑мышечных заболеваний как с мягким, так и с тяжелым течением, развитие которых обсуловлено мутациями в гене DMD, кодирующем белок дисторфин.
Тяжесть течения заболевания определяется типом мутации и функциональным доменом, в котором она локализуется.
Мягкое течение
Характеризуется бессимптомным повышением сывороточной креатинфосфокиназы и мышечными судорогами с миоглобинурией.
Тяжелое течение
Проявляется развитием классических синдромов, включающих в себя прогрессирующую мышечную дистрофию Дюшенна (ПМДД), прогрессирующую мышечную дистрофию Беккера (ПМДБ) и DMD‑ассоциированную дилатационную кардиомиопатию.
Заболевание характеризуется прогрессирующей слабостью проксимальных мышц, вызванной дегенерацией мышечных волокон. Дегенерация возникает в результате нарушения устойчивости и эластичности мышечных волокон при сокращениях. По мере развития заболевания мышечное волокно практически полностью разрушается и замещается соединительной тканью, что приводит к псевдогипертрофии мышц — увеличению их объема при утрате или значительном ослаблении функциональных возможностей. МДД входит в перечень наиболее распространенных Х-сцепленных заболеваний [1].
Прогрессирующая мышечная дистрофия, как правило, начинает проявляться с повышенной утомляемости и слабости мышц нижних конечностей.
Дифференциальный диагноз
Дистрофин
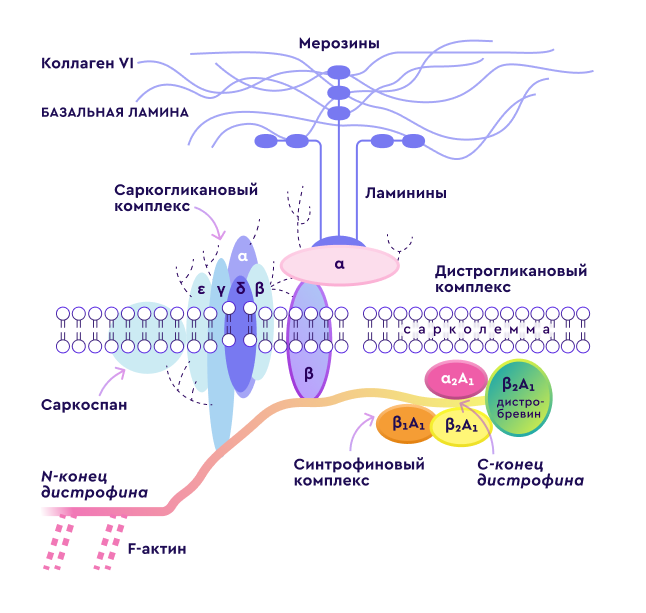
Дистрофин является мембранным белком, а дистрофин-ассоциированный комплекс представляет собой наиболее важный элемент мышечного цитоскелета, который обеспечивает взаимодействие внутренних и внешних структур клетки, участвует в регуляции уровня кальция в мышце и передаче импульсов через мембрану мышечного волокна.
Дистрофин находится, главным образом, в мышечных клетках и некоторых нейронах. В норме в его функции входит обеспечение эластичности и устойчивости мышечного волокна при сокращении.
При отсутствии дистрофина мембрана клетки разрушается и, как следствие, мышечное волокно разрушается и замещается соединительной тканью, что приводит к значительному ослаблению функциональных возможностей.
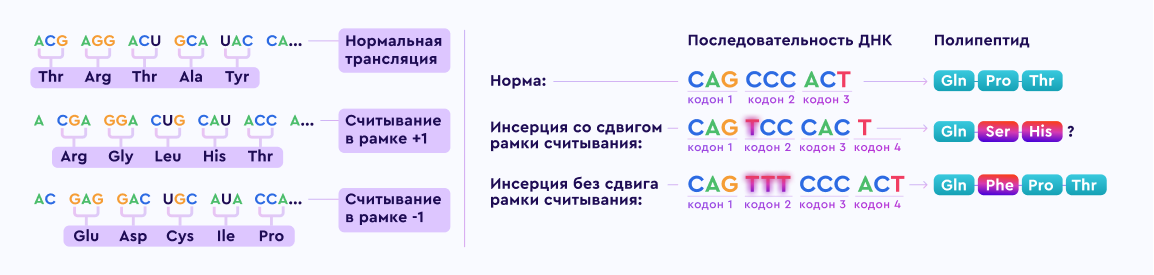
79%от общего числа мутаций составляют крупные делеции и дупликации.
21%составляют небольшие изменения, включающие однонуклеотидные замены, небольшие вставки и делеции, а также мутации сайтов сплайсинга, причем миссенс‑варианты не характерны для ПМДД/Б [3].
Особенности клинических проявлений связывают с типом мутации в гене дистрофина:
При ПМДД
Делеции в гене в большинстве случаев приводят к сдвигу рамки считывания и преждевременному окончанию считывания информации о построении белка, в результате дистрофин не образуется.
При ПМДБ
Структурные нарушения гена не нарушают рамку считывания, в результате образуется дефектный, функционально неполноценный белок.
При DMD-ассоциированной дилатационной кардиомиопатии
Функционально активный дистрофин отсутствует в миокарде, но присутствует в скелетной мускулатуре, поскольку при данном типе дистрофинопатий патогенные варианты приводят к различным тканеспецифической транскрипции или альтернативному сплайсингу в сердечной мыщце и в скелетной [Ferlini et al 1999, Neri et al 2007].
Прогрессирующая мышечная дистрофия Дюшенна
Является более распространенной формой данного заболевания. Первые признаки появляются в раннем возрасте (1‑5 лет) и характеризуются задержкой моторного развития. При начале ходьбы отмечаются частые падения, неловкость и утомляемость, трудности при подъеме по лестнице, беге и прыжках. Пациенты сохраняют способность к ходьбе до 10‑12 лет. Кардиомиопатия развивается практически у всех пациентов с ПМДД после 18 лет.
Заболевание быстро прогрессирует и гибель, как правило, наступает до 30‑летнего возраста в результате респираторных осложнений и прогрессии дилатационной кардиомиопатии.
Прогрессирующая мышечная дистрофия Беккера
Как правило, характеризуется более поздней манифестацией и мягкими клиническими проявлениями. Клинические признаки начинают проявляться примерно в интервале от 10 до 20 лет. Заболевание прогрессирует достаточно медленно, в большинстве случаев, пациент утрачивает способность к передвижению без инвалидной коляски не ранее 40‑летнего возраста.
Не смотря на более мягкий нервно-мышечный фенотип, сердечная недостаточность является наиболее частой причиной смерти при ПМДБ (средний возраст смерти 40‑50 лет).
DMD-ассоциированная дилатационная кардиомиопатия
По данным источников, мутации в гене DMD могут стать причиной не только ПМДД/Б, но и дилатационой кардиомиопатии 3B [2,6], которая является заболеванием сердечной мышцы и приводит к дисфункции желудочков и, как следствие, сердечной недостаточности. Женщины, гетерозиготные по патогенному варианту находятся в группе высокого риска развития данной патологии.
1:5000новорожденных мальчиков
Прогрессирующая мышечная дистрофия Дюшенна [4]
1:20-25000
Прогрессирующая мышечная дистрофия Беккера [9]
Xp21.2–p21.1Ген дистрофина локализован на коротком плече Х хромосомы (Xp21.2-p21.1) и является одним из самых протяженных в человеческом геноме (более 2Mb, содержит 79 экзонов).
Дистрофинопатии по типу наследования являются Х-сцепленными рецессивными заболеваниями. Пенетрантность дистрофинопатий полная у гемизиготных мальчиков; у гетерозиготных девочек-носителей патогенного варианта она варьирует.
Если женщина является носительницей гетерозиготной патогенной мутации в гене DMD, вероятности передачи мутации:
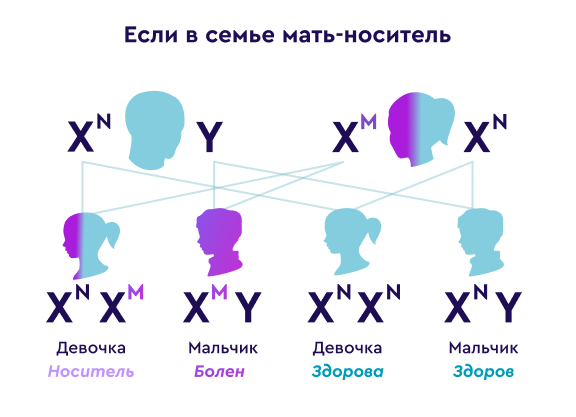
При беременности мальчиком — 50%Мутация будет передана сыну и он будет болен ПМДД/Б.
Все мальчики, унаследовавшие от матери патогенный вариант, будут больны.
При беременности девочкой — 50%Дочь унаследует данную мутацию и станет носительницей дефектной копии гена DMD.
Девочки, унаследовавшие патогенный вариант могут быть как бессимптомными носителями патогенной мутации в гетерозиготном состоянии, так и иметь клинические проявления классической дистрофинопатии.
Условия возникновения
При этом идентификация гетерозиготных носителей женского пола важна для клинического мониторинга кардиологических патологий у таких пациенток.
Больные ПМДД мужского пола, как правило, не успевают оставить потомство из-за ранней гибели; пациенты с ПМДБ и DMD-ассоциированной дилатационной кардиомиопатией могут иметь потомство: все их дочери будут гетерозиготными носителями патогенного варианта и ни один из сыновей не унаследует патогенный вариант.
Выявление носительства мутации гена DMD и планирование семьи с учетом риска рождения больного ребенка — самый эффективный способ профилактики дистрофинопатий.
Отец больного мальчика, как правило, не нуждается в молекулярно-генетическом тестировании, поскольку если он здоров, то он не может быть гемизиготным по патогенному варианту.
Остаточный риск — это вероятность рождения мальчика с дистрофинопатией, даже если при исследовании гена DMD у матери носительства патогенного варианта в лейкоцитарной ДНК не обнаружено.
Существование остаточного риска обусловлено тем, что в 15-20% случаев развитие мышечной дистрофии происходит из-за мутации, возникшей de novo (наиболее вероятен гонадный мозаицизм) [5]. В таком случае все братья пробанда-мальчика также имеют повышенный риск унаследовать патогенный вариант и при последующих беременностях целесообразно проведение пренатальной диагностики.
Прогрессирующая мышечная дистрофия Дюшенна:
Прогрессирующая мышечная дистрофия Беккера:
DMD-ассоциированная дилатационная кардиомиопатия:
Методы молекулярно-генетической диагностики
Подход к молекулярно-генетической диагностике дистрофинопатий заключается в последовательном проведении следующих этапов:
1. Исследование гена DMD
Поскольку большинство патогенных вариантов представляют собой делеции/дупликации одного и более экзонов, то целесообразно начать с анализа вариации числа копий (MLPA, хромосомный микроматричный анализ экзонного уровня). Если патогенный вариант не обнаружился, то следующим шагом проводится секвенирование гена DMD (с учетом протяженности гена, возможно начать поиск с «горячих точек»).
2. Исследование панели генов
Поиск мутаций, которые характерны для нервно-мышечных заболеваний, в том числе со сходными клиническими проявлениями (см. Дифференциальный диагноз).
3. Расширенный генетический анализ
Полноэкзомное или полногеномное секвенирование может быть проведен в случае нетипичных клинических проявлений, с целью уточнения диагноза и установления возможных причинных находок в других генах.
Как правило, исследование проводится с учетом информации о ранее выявленных патогенных вариантах в гене DMD у членов семьи.
Биологические образцы, пригодные для проведения молекулярно-генетического исследования:
Цитируемая литература:
1. Marina Basta, Ashish M. Pandya. Genetics, X-Linked Inheritance. Treasure Island. 2021 Jan; PMID: 32491315
2. https://www.omim.org/entry/302045
3. Hum Mutat. The TREAT-NMD DMD Global Database: Analysis of More than 7,000 Duchenne Muscular Dystrophy Mutations. Human Mutation. 2015 Apr; 36(4): 395–402.
4. Eppie M Yiu, Andrew J Kornberg. Duchenne muscular dystrophy. J Paediatr Child Health 2015 Aug; 51(8):759-64
5. «Метод молекулярно-генетического тестирования на носительство делеций и дубликаций экзонов гена DMD». Авторы: к.м.н., доцент Вильчук К.У., к.б.н. Гусина Н.Б., к.м.н. Гусина А.А., Мясников С.О., 2016.
6. Nakamura A. X-Linked Dilated Cardiomyopathy: A Cardiospecific Phenotype of Dystrophinopathy. Pharmaceuticals (Basel) 2015; 8:303–320.
7. Maria Sofia Falzarano, Chiara Scotton, Chiara Passarelli, Alessandra Ferlini. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules. 2015 Oct 7; 20(10):18168-84.
8. Van Westering T.L.E., Betts C.A., Wood M.J.A. Current understanding of molecular pathology and treatment of cardiomyopathy in Duchenne Muscular Dystrophy. Molecules. 2015
9. Иванов В.И., Барышникова Н.В., Билева Д.С., Дадали Е.Л., Генетика. 2006

Многолетний опыт работы в области генетики, лабораторной диагностики и биоинформатики

Все данные строго конфиденциальны и не могут быть переданы третьим лицам

Особый контроль на каждом этапе проведения исследования

Результаты в короткие сроки

Доставка биоматериала по всей России
- наличия делеции Х-хромосомы в области Xp21.2;
- однородительской дисомии по Х-хромосоме;
- компаунд-гетерозиготном состоянии двух патогенных вариантов;
- неслучайной Х-инактивации